

Της Ζένιας Κουφοπαντελή,
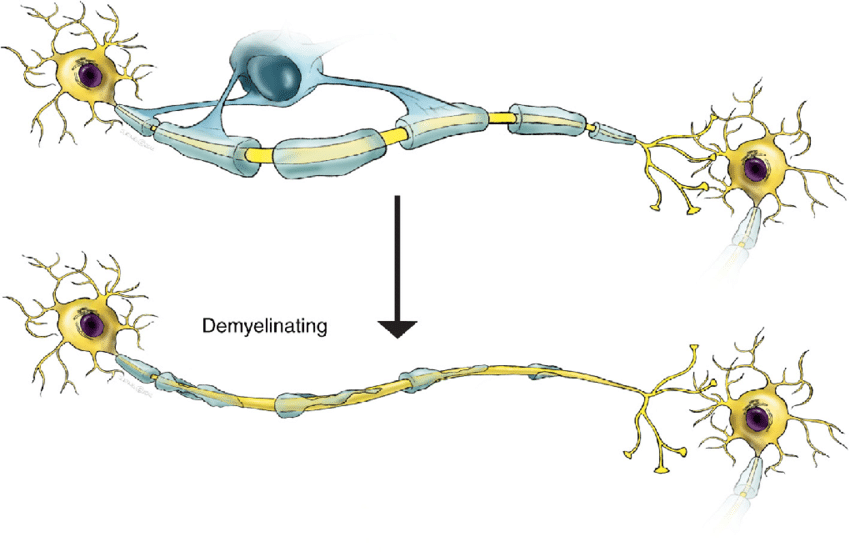
Η νόσος Charcot-Marie-Tooth (CMT) αποτελεί μία από τις πιο κοινές κληρονομικές νευρολογικές διαταραχές, και πήρε το όνομά της από τους τρεις γιατρούς που την περιέγραψαν για πρώτη φορά το 1886, τους Jean-Martin Charcot και Pierre Marie από τη Γαλλία, και τον Howard Henry Tooth από το Ηνωμένο Βασίλειο. Ο συνολικός εκτιμώμενος επιπολασμός της CMT είναι περίπου 19 περιπτώσεις ανά 100.000. Η νόσος εμφανίζεται λόγω βλάβης στα περιφερικά νεύρα, τα οποία μεταδίδουν σήματα από τον εγκέφαλο και τον νωτιαίο μυελό στους μυς και μεταβιβάζουν αισθήσεις, όπως ο πόνος και η αφή, από το υπόλοιπο σώμα στο Κεντρικό Νευρικό Σύστημα. Η βλάβη αυτή προκαλείται από μεταλλάξεις στα γονίδια υπεύθυνα για τη δημιουργία και τη διατήρηση της μυελίνης που είναι απαραίτητη για την ταχεία αγωγή των νευρικών ώσεων και περιβάλλει τους άξονες των νευρικών κυττάρων παρέχοντας προστασία σε αυτά.
Περισσότερα από 40 γονίδια έχει βρεθεί να διαδραματίζουν ρόλο στην εμφάνιση της νόσου, με κάθε γονίδιο να συνδέεται με έναν ή περισσότερους τύπους της νόσου. Επιπλέον, πολλαπλά γονίδια μπορεί να συνδέονται με έναν τύπο CMT, με περισσότερες από τις μισές περιπτώσεις να προκαλούνται από έναν διπλασιασμό του γονιδίου PMP22 στο χρωμόσωμα 17. Παρά το ότι διαφορετικές πρωτεΐνες είναι μεταλλαγμένες στις διάφορες μορφές της πάθησης, όλες οι μεταλλάξεις επηρεάζουν κυρίως τη φυσιολογική λειτουργία των περιφερικών νεύρων. Οι μεταλλάξεις που αφορούν στην ελαττωματική παραγωγή μυελίνης προκαλούν δυσλειτουργία της επικάλυψης των νευραξόνων, η οποία επιβραδύνει την αγωγή των νευρικών ώσεων, ενώ άλλες μεταλλάξεις περιορίζουν τη λειτουργία των αξόνων και προκαλούν αξονική απώλεια. Άλλοι τύποι CMT κληρονομούνται κατά τρόπο συνδεδεμένο με το Χ, δηλαδή εξαρτώνται από τα χρωμοσώματα που καθορίζουν το φύλο του ατόμου.
Γονιδιακό υπόβαθρο
Η νόσος διακρίνεται σε τέσσερις κύριες μορφές που σχετίζονται με τέσσερα διαφορετικά γονίδια, οι οποίες αντιπροσωπεύουν το 90% όλων των περιπτώσεων CMT:
- Η CMT1A, που οφείλεται στον διπλασιασμό του γονιδίου PMP22, είναι η πιο συχνή διαταραχή CMT με αυτοσωματικό επικρατή τύπο κληρονομικότητας
- Η CMTX1, που οφείλεται σε μεταλλάξεις του GJB1, γονιδίου που συνδέεται με το Χ
- Η CMT1B, που οφείλεται σε μεταλλάξεις του αυτοσωμικού γονιδίου MPZ
- Η CMT2A, που οφείλεται σε μεταλλάξεις του γονιδίου MFN2 έχει επίσης αυτοσωμική
επικρατούσα κληρονομικότητα
Συμπτωματολογία
Τα πρώτα σημάδια εμφανίζονται συνήθως πριν από την ηλικία των 20 ετών, αλλά ενδέχεται να εμφανιστούν και αργότερα. Οι κλασικές κλινικές εκδηλώσεις αφορούν σε κινητική δυσχέρεια και διαταραχές της αισθητικότητας, μυϊκή ατροφία και παραμορφώσεις των οστών, συμπεριλαμβανομένης της εμφάνισης «κοίλου ποδιού». Η πάθηση χαρακτηρίζεται από την εμφάνιση σημαντικής κλινικής ετερογένειας με τη σοβαρότητα της νόσου είναι μεταβάλλεται μεταξύ των ασθενών, κυμαινόμενη από ασυμπτωματικές μορφές στους ενήλικες έως πολύ σοβαρές μορφές στα παιδιά. Η επιδείνωση είναι σταδιακή με συμπτώματα που περιλαμβάνουν:
- Αδυναμία ή παράλυση των μυών του ποδιού και του κάτω μέρους του ποδιού, η οποία
μπορεί να προκαλέσει δυσκολία στην ανύψωση του ποδιού - Δυσκολίες ισορροπίας
- Παραμορφώσεις των ποδιών
- Απώλεια σημαντικού ποσοστού του μυϊκού όγκου στα κάτω άκρα
- Μειωμένη ικανότητα αίσθησης της θερμότητας, του κρύου και της αφής
Προς το παρόν, δεν έχει βρεθεί κάποιο φάρμακο ικανό να καταπολεμήσει πλήρως τη νόσο, ωστόσο έχουν βρεθεί ποικίλες ουσίες που ενδέχεται να εμφανίζουν θεραπευτική δράση, όπως το ασκορβικό οξύ, η σορβιτόλη, η ναλτρεξόνη, η νευροτροφίνη 3 και η μπακλοφαίνη. Η περίθαλψη των ασθενών βασίζεται στην αποκατάσταση μέσω εργοθεραπείας, η οποία στοχεύει στη διατήρηση των ικανοτήτων των ασθενών, σε συνδυασμό με ψυχολογική υποστήριξη. Η σωματική άσκηση πρέπει να προσαρμόζεται στις ανάγκες κάθε ασθενούς και να περιλαμβάνει μυϊκή ενδυνάμωση, αερόβια άσκηση και χαλάρωση. Έχει αποδειχθεί ότι οι ήπιας έως μέτριας έντασης ασκήσεις ενισχύουν τους μυς των άνω και των κάτω άκρων και συμβάλλουν στην ενδυνάμωση των άκρων στο σύνολό τους. Τέλος, μία άλλη θεραπευτική παρέμβαση περιλαμβάνει τη διενέργεια χειρουργικής επέμβασης, μπορεί να πραγματοποιηθεί για την επιδιόρθωση παραμορφώσεων των άκρων. Η χειρουργική επέμβαση, ωστόσο, ενδείκνυται ως τελευταία επιλογή όταν η φυσικοθεραπεία ή η εργοθεραπεία έχουν αποτύχει.
ΕΝΔΕΙΚΤΙΚΕΣ ΠΗΓΕΣ
- Charcot-Marie-Tooth Disease, National Institute of Neurological Disorders and Stroke. Διαθέσιμο εδώ
- Charcot-Marie-Tooth disease, Mayo Clinic. Διαθέσιμο εδώ
- Treatment of Charcot-Marie-Tooth neuropathies, Science Direct. Διαθέσιμο εδώ
- Charcot-Marie-Tooth Disease (CMT), MDA. Διαθέσιμο εδώ

