

Της Ταξιαρχίας Κοζπή,
Όταν η κόρη της Pearl Buck, η Carol, έγινε τριών ετών, άρχισε να φαίνεται ότι δεν ήταν σαν τα υπόλοιπα παιδιά της ηλικίας της. Μεταφέρθηκε στη Μινεσότα των Η.Π.Α από τους γονείς της, έτσι ώστε να υποβληθεί στις απαραίτητες εξετάσεις. Οι γιατροί είπαν πως ο εγκέφαλος του παιδιού δεν αναπτυσσόταν κανονικά. H ασθένεια, από την οποία έπασχε η Carol, ανιχνεύτηκε από τους επιστήμονες, όταν το κορίτσι έγινε τριάντα ετών. Η νόσος θα μπορούσε να καταπολεμηθεί με την εφαρμογή ενός ειδικού διαιτολογίου. Δυστυχώς, όμως, η Carol ήταν ήδη σε μεγάλη ηλικία για να αντιμετωπιστεί το πρόβλημά της. Δεκαέξι γράμματα, επτά συλλαβές: φαι-νυλ-κε-το-νου-ρί-α. Από αυτήν την πάθηση έπασχε η Carol, ενώ υπολογίζεται ότι από αυτήν νοσούν και 1:10.000 νεογνά σε Ευρώπη και Αμερική.
H φαινυλκετονουρία αποτελεί κληρονομικό νόσημα της ομάδας των υπερφαινυλαλανιναιμιών, με χαρακτηριστικό την ενδογενή αδυναμία μεταβολισμού του αμινοξέος φαινυλαλανίνη προς τυροσίνη. Ας πάρουμε τα πράγματα, όμως, από την αρχή. Ο μεταβολισμός είναι το σύνολο των βιοχημικών αντιδράσεων που λαμβάνουν χώρα σε όλα τα κύτταρα του οργανισμού μας. Ο οργανισμός μας χρησιμοποιεί ποικίλα θρεπτικά συστατικά ανάλογα με τις ενεργειακές του ανάγκες. Ορισμένες φορές, όμως, δημιουργούνται προβλήματα, τα οποία οδηγούν στην εμφάνιση νοσημάτων. Τα μεταβολικά νοσήματα, λοιπόν, αφορούν τον μεταβολισμό των αμινοξέων, των λιπαρών οξέων και των υδατανθράκων. Τα κληρονομικά μεταβολικά νοσήματα ή αλλιώς οι ενδογενείς διαταραχές του μεταβολισμού, όπως η φαινυλκετονουρία, είναι αποτέλεσμα γονιδιακών μεταλλάξεων. Οι γονιδιακές μεταλλάξεις αφορούν αλλαγές στη σειρά των βάσεων ενός γονιδίου. Το γενετικό μας υλικό, το DNA, αποτελείται από γονίδια, δηλαδή μικρά τμήματα DNA, υπεύθυνα για τον έλεγχο γνωρισμάτων και χαρακτηριστικών. Με άλλα λόγια, το γονίδιο είναι η βασική μονάδα της κληρονομικότητας.

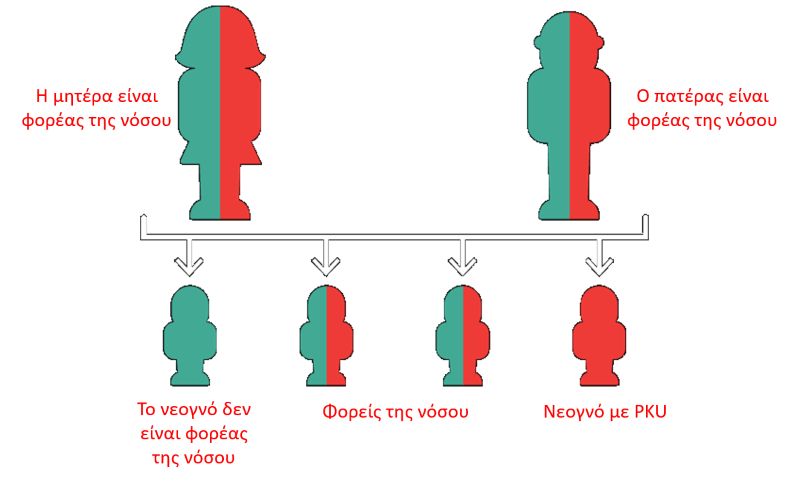
Η φαινυλκετονουρία κληρονομείται με αυτοσωμικό υπολειπόμενο τρόπο (οι αυτοσωμικές υπολειπόμενες ασθένειες εκδηλώνονται στα παιδιά που έχουν κληρονομήσει ένα παθολογικό γονίδιο από κάθε γονέα). Η ασθένεια είναι αποτέλεσμα της ανεπάρκειας ή της έλλειψης του ηπατικού ενζύμου, υδροξυλάση της φαινυλαλανίνης (PAH), που είναι απαραίτητο για τη μετατροπή της φαινυλαλανίνης σε τυροσίνη. Όλοι μας έχουμε στο DNA μας δύο γονίδια, το προϊόν της έκφρασης των οποίων είναι το ένζυμο υδροξυλάση της φαινυλαλανίνης (PAH). Στους φορείς της νόσου, το ένα από αυτά τα γονίδια είναι ελαττωματικό και οδηγεί στην παραγωγή ενζύμου με μειωμένη ή απούσα δραστικότητα. Παρ’ όλα αυτά, οι φορείς δεν εμφανίζουν τη νόσο, διότι το άλλο γονίδιο είναι φυσιολογικό και επικρατεί του μη φυσιολογικού. Προκειμένου ένα νεογνό να εκδηλώσει την ασθένεια, χρειάζεται να λάβει και από τους δύο γονείς το παθολογικό γονίδιο, δηλαδή πρέπει και οι δύο γονείς να είναι φορείς της νόσου.
Η συσσώρευση φαινυλαλανίνης στο αίμα και σε διάφορους ιστούς, συμπεριλαμβανομένου και του εγκεφάλου, προκαλεί σοβαρές νευρολογικές διαταραχές, όπως διανοητική καθυστέρηση. Η σοβαρότητα των φαινοτύπων της νόσου εξαρτάται από τον βαθμό της ανεπάρκειας του ενζύμου PAH, αλλά και του συνενζύμου της, της τετραϋδροβιοπτερίνης (BH4), που ρυθμίζουν την αποδόμηση της φαινυλαλανίνης. Η κλινική εικόνα των ασθενών, λοιπόν, ποικίλλει αρκετά. Η νόσος προκαλεί απομυελινώσεις, ενώ εμφανίζονται διαταραχές, όπως σπασμοί, αταξία, προβλήματα συμπεριφοράς και διανοητική καθυστέρηση. Έχουν διερευνηθεί τρεις κύριοι τύποι φαινυλκετονουρίας.
Στην πρώτη περίπτωση, περιλαμβάνονται παιδιά που εμφανίζουν μια καλοήθη κατάσταση, γνωστή ως υπερφαινυλαλανιναιμία. Σε αυτή την περίπτωση, δεν είναι αναγκαία η θεραπεία, εφόσον δεν υπάρχει κίνδυνος εμφάνισης διανοητικής καθυστέρησης. Αποτελεί μια απλή παροδική κατάσταση, που αποκαθίσταται τον πρώτο μήνα ζωής του νεογνού και δεν εμφανίζονται κλινικά συμπτώματα. Στη δεύτερη κατηγορία, περιλαμβάνονται παιδιά με μια απλή επίμονη υπερφαινυλαλανιναιμία. Υπάρχει μικρή ανεπάρκεια του ενζύμου της υδροξυλάσης της φαινυλαλανίνης. Οι τιμές φαινυλαλανίνης αίματος είναι 4 – 10 mg/dl και η κλινική εικόνα είναι φυσιολογική. Η τρίτη κατηγορία είναι η κλασική φαινυλκετονουρία, στην οποία παρουσιάζεται μέτρια έως μεγάλη ανεπάρκεια της υδροξυλάσης της φαινυλαλανίνης. Σε αυτόν τον τύπο της νόσου, η κλινική εικόνα περιλαμβάνει σπασμούς, μικροκεφαλία, αδυναμία συγκέντρωσης, δυσάρεστη οσμή ούρων και διανοητική καθυστέρηση.
Η φαινυλκετονουρία παρατηρήθηκε και μελετήθηκε για πρώτη φορά το 1934 από τον Νορβηγό Ιατρό Φέλλινγκ, ο οποίος ανίχνευσε μία ουσία, τη φαινυλοακετόνη, στα ούρα των ασθενών. Το 1951, οι Woolf & Vulliamy ήταν οι πρώτοι που ανακάλυψαν πως τα επίπεδα της φαινυλαλανίνης μπορούν να μειωθούν, αν διακοπεί καθολικά η διατροφική πρόσληψή της. Αργότερα, το 1960 δημιουργήθηκαν τα πρώτα προγράμματα νεογνικού ελέγχου στις ΗΠΑ και ακολουθήσαν και οι άλλες χώρες.
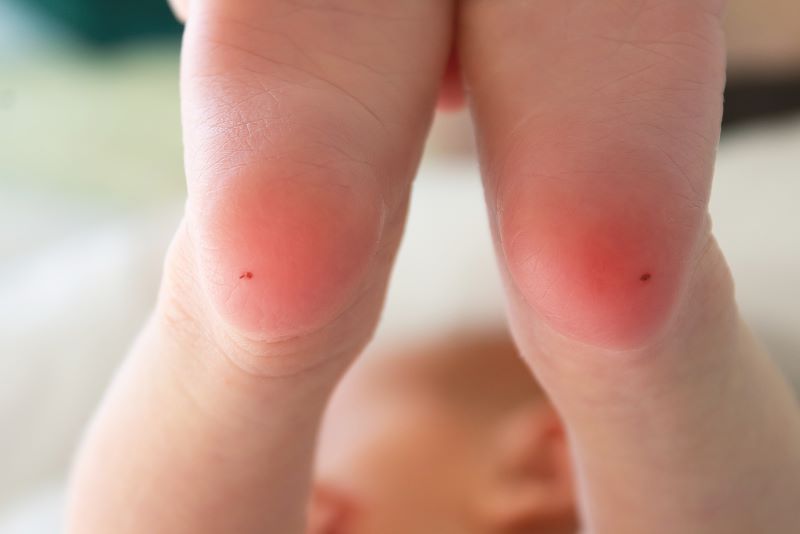
Η διάγνωση της νόσου μπορεί να διεξαχθεί με διάφορες μεθόδους. Κατά τον προγεννητικό έλεγχο, πραγματοποιείται test γενετικού υλικού DNA. Το ίδιο test διενεργείται και για την εύρεση των μελών μίας οικογένειας, που φέρει το μεταλλαγμένο γονίδιο της PKU. Στα νεογέννητα, ο έλεγχος αυτός γίνεται εντός των πρώτων ημερών ζωής, εκτός, όμως, του πρώτου 24ώρου, όπου λαμβάνονται κάποιες σταγόνες αίματος από τη φτέρνα του βρέφους. Στη συνέχεια, ελέγχονται τα επίπεδα της φαινυλαλανίνης και σε περίπτωση που αυτά διαφέρουν από το φυσιολογικό, πραγματοποιούνται και άλλες ειδικότερες εξετάσεις για την ανίχνευση της PKU.
Μία άλλη μέθοδος είναι το test Guthrie. Το test Guthrie αποτελεί και αυτό test αίματος των νεογνών (βιοχημική μέθοδος ανίχνευσης), όπου το δείγμα λαμβάνεται σε ειδική διηθητική κάρτα από τα μαιευτήρια, πριν πάρει εξιτήριο το βρέφος για το σπίτι, συνήθως την 3η με 5η μέρα από τη γέννηση. Με μέσο μια ειδική βελόνα, οι ειδικοί λαμβάνουν σταγόνες αίματος από το πόδι του βρέφους. Το δείγμα αποστέλλεται ταχυδρομικά στο Ινστιτούτο Υγείας Παιδιού, όπου γίνεται έλεγχος για τρία νοσήματα, μεταξύ αυτών και για τη φαινυλκετονουρία. Το συγκεκριμένο test καλό είναι να αποφεύγεται, όταν το νεογέννητο λαμβάνει αντιβιοτικά, καθώς μπορεί να οδηγήσει σε ψευδώς αρνητικό αποτέλεσμα. Λόγω του παραπάνω γεγονότος, είναι προτιμότερο το test γενετικού υλικού DNA, αφού δεν επηρεάζεται από τη λήψη αντιβιοτικών.
Τέλος, υπάρχουν και οι εξετάσεις για την ανίχνευση φαινυλο – πυροσταφυλικού οξέος στα ούρα. Η ανίχνευση της συγκεκριμένης ουσίας επιβεβαιώνει την ύπαρξη της ασθένειας και συνεισφέρει στον σχεδιασμό κατάλληλου διαιτολογίου. Σύμφωνα με το Medical Research Council, η διάγνωση και η θεραπεία πρέπει να ξεκινάνε στις 20 πρώτες μέρες της ζωής του ασθενούς. Σε διαφορετική περίπτωση, τα συμπτώματα της ασθένειας είναι μη αναστρέψιμα.
Απώτερος στόχος της θεραπείας είναι η διατήρηση των επιπέδων της φαινυλαλανίνης σε επιτρεπτά όρια. Πρέπει να τονιστεί ότι η φαινυλαλανίνη δεν παράγεται από τον οργανισμό, αλλά εισέρχεται σε αυτόν μέσω της διατροφής. Η ελεγχόμενη, λοιπόν, χορήγηση φαινυλαλανίνης μέσω της διατροφής αποτελεί τον ακρογωνιαίο λίθο για την αντιμετώπιση της νόσου. Η θεραπευτική αυτή λύση προτάθηκε έναν χρόνο μετά από την ανακάλυψη της ασθένειας, από τον Γερμανό ιατρό Bickel. Οι επιστήμονες ήλπιζαν ότι η χορήγηση της υδροξυλάσης της φαινυλαλανίνης (PAH) θα αποτελούσε, επίσης, μέθοδο θεραπείας, αλλά αυτό αποδείχθηκε ανέφικτο. Σημαντικό να αναφερθεί ότι εκτός από τη δίαιτα χαμηλή σε φαινυλαλανίνη, απαραίτητη είναι και η επαρκής πρόσληψη τυροσίνης. Αρχικά, ένα παιδί με φαινυλκετονουρία θα πρέπει να λαμβάνει 200 – 500 mg/ημέρα φαινυλαλανίνης μέχρι και την ηλικία των δώδεκα, καθώς και 25 – 85 mg/κιλό σωματικού βάρους/ημέρα τυροσίνης. Η καταλληλότερη μέθοδος σίτισης των νεογνών κρίνεται πως είναι η συνδυασμένη λήψη μητρικού γάλακτος (στο οποίο περιλαμβάνεται λιγότερη φαινυλαλανίνη από ό,τι στο αγελαδινό) και ειδικών φόρμουλων «ελεύθερων» σε φαινυλαλανίνη.

Η διατροφή PKU πρέπει να περιλαμβάνει λιγότερη πρωτεΐνη, περισσότερους υδατάνθρακες και λιγότερο συνολικό λίπος, σε σύγκριση με μια διατροφή ενός υγιούς ατόμου. Η διατροφή αυτή, λοιπόν, στερείται κρέατος, πουλερικών, ψαριών, καρπών με κέλυφος και γαλακτοκομικών προϊόντων. Τα επίπεδα λιπιδίων στο αίμα θα πρέπει να παρακολουθούνται, η πρόσληψη φυτικών ελαίων πλούσιων σε λινολενικό οξύ, όπως το canola ή soybeans oil, θα πρέπει να ενθαρρύνεται και τα ισορροπημένα συμπληρώματα ω-3 και ω-6 LC-PUFA να παρέχονται, όπως απαιτείται για όλα τα άτομα με PKU. Τρόφιμα που περιέχουν ως πρόσθετη ύλη την ασπαρτάμη δε μπορούν να καταναλωθούν από τους πάσχοντες, γιατί αυτή διασπάται σε ασπαρτικό οξύ, φαινυλαλανίνη και μικρές ποσότητες μεθανόλης. Η ασπαρτάμη χρησιμοποιείται ως υποκατάστατο της ζάχαρης σε τρόφιμα χαμηλής θερμιδικής αξίας. Το πιο διαδεδομένο παράδειγμα είναι τα light αναψυκτικά. Τέλος, οι πάσχοντες από φαινυλκετονουρία χρειάζεται να μάθουν να αναγιγνώσκουν προσεκτικά τις ετικέτες των τροφίμων. Καθίσταται υποχρεωτική η αναγραφή της περιεκτικότητας της φαινυλαλανίνης και της ασπαρτάμης σε όλες τις συσκευασίες τροφίμων.
Ο υπολογισμός των επιπέδων φαινυλαλανίνης στο αίμα είναι καθοριστικής σημασίας για την πορεία και τις τυχόν μεταβολές του διαιτολογίου. Ωστόσο, λοιμώξεις, μετεγχειρητικό στρες, χαμηλή ενεργειακή πρόσληψη και άλλες καταστάσεις, μπορεί να αυξήσουν τη συγκέντρωση φαινυλαλανίνης στο αίμα, δυσχεραίνοντας την εκτίμηση. Παλαιότερα, η εφαρμογή ενός αυστηρού διαιτολογίου ως προς την περιεκτικότητά του σε φαινυλαλανίνη, αποτελούσε μέριμνα μέχρι την παιδική ηλικία, αφού πίστευαν πως η ολοκλήρωση της ανάπτυξης του κεντρικού νευρικού συστήματος ελάττωνε την ανάγκη για μετέπειτα αυστηρή συμμόρφωση με την ειδική διατροφή. Αποτελέσματα σύγχρονων μελετών καταδεικνύουν πως η εφαρμογή ενός διαιτολογίου χαμηλής περιεκτικότητας σε φαινυλαλανίνη είναι αναγκαία εφόρου ζωής στους νοσούντες και συνδέεται με αυξημένη γνωσιακή απόδοση και χαμηλό ρίσκο για νευρολογικές ανωμαλίες.
ΕΝΔΕΙΚΤΙΚΕΣ ΠΗΓΕΣ
- Τεστ αίματος Guthrie, pediatros-thes.gr. Διαθέσιμο εδώ.
- Psychiatric and Cognitive Aspects of Phenylketonuria: The Limitations of Diet and Promise of New Treatments, ncbi.nlm.nih.gov. Διαθέσιμο εδώ.
- Η σημασία της διατροφής στην φαινυλκετονουρία, dkdiet.gr. Διαθέσιμο εδώ.
- Μαρία Καλογεράκου, Ιατρική Σχολή Εθνικού και Καποδιστριακού Πανεπιστημίου Αθηνών, Μελέτη της συσχέτισης της φαινυλκετονουρίας και γαλακτοζαιμίας με τη σκελετική υγεία: Μια εργαστηριακή και κλινική περίπτωση. Διαθέσιμο εδώ

