

Της Μαρίας Δήμα,
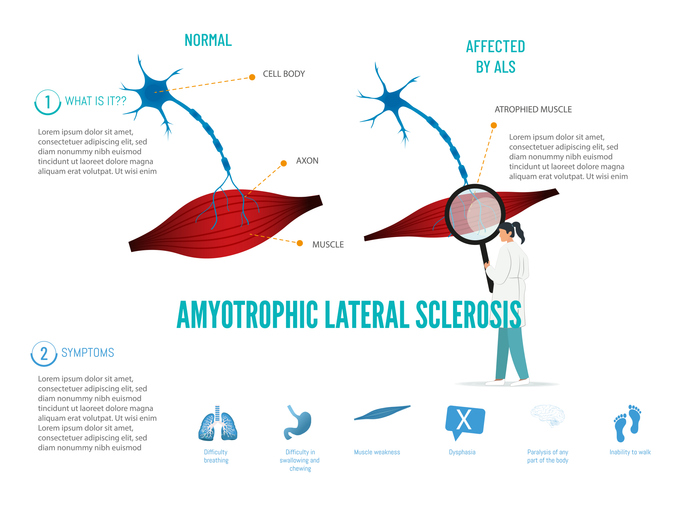
Η πλάγια αμυοτροφική σκλήρυνση (ALS) ή νόσος του κινητικού νευρώνα ή αλλιώς νόσος του Lou Gehrig, είναι μια σπάνια, προοδευτική νευροεκφυλιστική νόσος που επηρεάζει τους κινητικούς νευρώνες του εγκεφάλου και του νωτιαίου μυελού. Χαρακτηρίζεται από σταδιακή απώλεια των εκούσιων μυϊκών κινήσεων, οδηγώντας τελικά σε παράλυση. Η ALS ανήκει στις νόσους του κινητικού νευρώνα και είναι ανίατη. Η επίπτωση της νόσου κυμαίνεται περίπου μεταξύ 1,5 και 3 περιπτώσεων ανά 100.000 άτομα ετησίως, με ελαφρώς υψηλότερη συχνότητα στους άνδρες σε σχέση με τις γυναίκες. Συνήθως εμφανίζεται σε άτομα ηλικίας 40-70 ετών, με τη μέση ηλικία διάγνωσης να είναι περίπου τα 55 έτη. Παρόλο που η νόσος μπορεί να προσβάλει οποιονδήποτε, ορισμένοι γενετικοί και περιβαλλοντικοί παράγοντες φαίνεται να αυξάνουν τον κίνδυνο εμφάνισής της.
Η ακριβής αιτία της ALS παραμένει ασαφής, αλλά υπάρχουν ενδείξεις ότι προκύπτει από ένα συνδυασμό γενετικών και περιβαλλοντικών παραγόντων. Περίπου το 5-10% των περιπτώσεων είναι κληρονομικές (οικογενειακή ALS) και συνδέονται με μεταλλάξεις σε γονίδια όπως το C9orf72, το SOD1, το TARDBP, και το FUS. Οι υπόλοιπες περιπτώσεις είναι σποραδικές, δεν έχουν σαφή γενετική προδιάθεση, αλλά πιθανοί παράγοντες κινδύνου περιλαμβάνουν την έκθεση σε τοξίνες, βαρέα μέταλλα, φυτοφάρμακα, ιούς, και τραυματισμούς στο κρανίο. Επιπλέον, διαταραχές στην πρωτεϊνική αναδίπλωση, καθώς και το οξειδωτικό στρες φαίνεται να διαδραματίζουν σημαντικό ρόλο στην παθολογία της νόσου.
Έχουν προταθεί τρεις θεωρίες που εξηγούν την παθογένεια της ALS. Η πρώτη είναι γνωστή και ως η υπόθεση του “dying forward”. Προτείνει ότι η ALS έχει φλοιϊκή προέλευση και περιλαμβάνει τους κινητικούς νευρώνες του φλοιού που συνδέονται με τους κινητικούς νευρώνες του νωτιαίου μυελού μέσω μονοσυναπτικών συνδέσεων. Η μετάδοση αυτή διαμεσολαβείται από τη γλουταμινική εξωτοξικότητα, οδηγώντας σε εκφύλιση των κινητικών νευρώνων κατά μήκος των αξόνων. Η δεύτερη θεωρίας (υπόθεση του “dying back”) υποστηρίζει ότι η δυσλειτουργία των κατώτερων κινητικών νευρώνων συμβαίνει νωρίς στην πορεία της νόσου, πιθανώς λόγω προέλευσής της στους μύες ή στη νευρομυϊκή σύναψη.
Εκεί, πραγματοποιείται ανάδρομη μεταφορά επιβλαβών παραγόντων στα κυτταρικά σώματα μέσω των αξόνων, προκαλώντας τοξικότητα — ένα φαινόμενο που μπορεί να σχετίζεται με τη δυσλειτουργία της αξοπλασματικής μεταφοράς (μεταφορά πρωτεϊνών, λιπιδίων, οργανιδίων κλπ στο κυτταρικό σώμα του νευρώνα διαμέσω της κυτταροπλασματικής του μεμβράνης). Η τρίτη και τελευταία θεωρία, είναι η υπόθεση της ανεξάρτητης εκφύλισης, η οποία αναφέρει ότι οι εκφυλιστικές αλλοιώσεις των φλοιονωτιαίων και κατώτερων κινητικών νευρώνων συμβαίνουν ανεξάρτητα και τυχαία, και πιστεύεται ότι εξαπλώνονται κατά μήκος των αντίστοιχων νευροανατομικών τους δομών.
Η ALS χαρακτηρίζεται από εκφυλισμό των ανώτερων και κατώτερων κινητικών νευρώνων, γεγονός που οδηγεί σε αδυναμία μετάδοσης των νευρικών σημάτων προς τους μύες. Οι ανώτεροι κινητικοί νευρώνες, που βρίσκονται στον εγκεφαλικό φλοιό, εκφυλίζονται πρώτοι, ακολουθούμενοι από τους κατώτερους κινητικούς νευρώνες του νωτιαίου μυελού. Αυτό έχει ως αποτέλεσμα την προοδευτική απώλεια μυϊκής δύναμης, ατροφία και σπαστικότητα. Παράλληλα, παρατηρείται συσσώρευση ελαττωματικών πρωτεϊνών στα νευρικά κύτταρα – με συχνότερη την πρωτεΐνη TDP-43 -, δυσλειτουργία των μιτοχονδρίων και φλεγμονώδεις αντιδράσεις, οι οποίες συμβάλλουν περαιτέρω στην καταστροφή των νευρικών κυττάρων.
Τα αρχικά συμπτώματα της ALS είναι ήπια και συχνά περνούν απαρατήρητα. Περιλαμβάνουν μυϊκή αδυναμία, κράμπες και μυϊκές συσπάσεις, συνήθως στα άκρα ή στη γλώσσα. Καθώς η νόσος εξελίσσεται, οι ασθενείς εμφανίζουν προοδευτική απώλεια κινητικότητας, ατροφία των μυών της μάσησης και κατάποσης, αδυναμία στήριξης του κορμού και του κεφαλιού, και τέλος, ατροφία των αναπνευστικών μυών. Σε ένα ποσοστό των ασθενών (30-50%), εμφανίζονται νοητικές και συμπεριφορικές διαταραχές, όπως απώλεια μνήμης, αφασία και αντικοινωνική συμπεριφορά, ενώ αξίζει να επισημανθεί, ότι στην πλειοψηφία των ατόμων με ALS, η όραση, η ακοή, η όσφρηση, η αφή και η γεύση διατηρούνται. Δυνητικά, η αδυναμία κατάποσης οδηγεί σε εισροφήσεις, γεγονός το οποίο αυξάνει τον κίνδυνο πνευμονίας από εισρόφηση. Λόγω αδυναμίας των μυών της μάσησης και κατάποσης, το άτομο υποσιτίζεται σημαντικά, ενώ σε προχωρημένο στάδιο, η αναπνευστική ανεπάρκεια αποτελεί την κύρια αιτία θανάτου. Το προσδόκιμο ζωής τυπικά δεν υπερβαίνει τα 5 χρόνια.

Η διάγνωση της νόσου, συνήθως τίθεται κλινικά με βάση τα συμπτώματα του ασθενούς. Λαμβάνεται πλήρες ατομικό και οικογενειακό ιστορικό αναμνηστικό, ενώ διενεργείται πλήρης νευρολογικός έλεγχος για την διαφοροδιάγνωση από άλλες παθήσεις του νευρικού και μυϊκού συστήματος. Τελικά, η διάγνωση της νόσου επιβεβαιώνεται με ένα ηλεκτρομυογράφημα.
Προς το παρόν, δεν υπάρχει θεραπεία που να σταματά την εξέλιξη της ALS, και η αντιμετώπισή της είναι κατά βάση συμπτωματική. Διενεργείται ένας συνδυασμός θεραπευτικών προσεγγίσεων, όπως φαρμακευτική αγωγή, φυσικοθεραπεία και εργοθεραπεία, διατροφική και αναπνευστική υποστήριξη, καθώς και βοηθητικές συσκευές για ομιλία και κίνηση. Ωστόσο, υπάρχουν φαρμακευτικές και υποστηρικτικές παρεμβάσεις που μπορούν να επιβραδύνουν την πρόοδο της νόσου και να βελτιώσουν την ποιότητα ζωής των ασθενών. Τα κύρια φάρμακα που χρησιμοποιούνται είναι το Riluzole, το οποίο μειώνει τη βλάβη των νευρώνων μέσω της αναστολής της απελευθέρωσης γλουταμινικού οξέος (ανασταλτικός νευροδιαβιβαστής), και το Edaravone, που δρα ως αντιοξειδωτικό. Οι υποστηρικτικές θεραπείες περιλαμβάνουν φυσικοθεραπεία, εργοθεραπεία, λογοθεραπεία και αναπνευστική υποστήριξη, όπως μη επεμβατικό αερισμό. Επιπλέον, η διατροφή παίζει σημαντικό ρόλο, καθώς η απώλεια βάρους επιδεινώνει την κατάσταση των ασθενών. Η έρευνα συνεχίζεται με έμφαση σε θεραπείες γονιδιακής θεραπείας, νευροπροστασίας και κυτταρικής αναγέννησης.
Η πλάγια αμυοτροφική σκλήρυνση είναι μια σοβαρή, ανίατη νόσος που επηρεάζει το νευρικό σύστημα και καταλήγει σε προοδευτική παράλυση. Παρότι οι αιτίες της δεν είναι πλήρως κατανοητές, οι επιστήμονες συνεχίζουν να ερευνούν πιθανούς μηχανισμούς και θεραπείες. Η έγκαιρη διάγνωση και η υποστηρικτική φροντίδα μπορούν να βελτιώσουν σημαντικά την ποιότητα ζωής των ασθενών, ενώ οι συνεχιζόμενες έρευνες δίνουν ελπίδα για πιο αποτελεσματικές θεραπευτικές παρεμβάσεις στο μέλλον.
ΕΝΔΕΙΚΤΙΚΕΣ ΠΗΓΕΣ
- ALS – Νόσος του κινητικού νευρώνα, Neurocenter.gr, διαθέσιμο εδώ
- Pathological mechanisms of amyotrophic lateral sclerosis, PubMed, διαθέσιμο εδώ
- Amyotrophic Lateral Sclerosis (ALS), Cleveland Clinic, διαθέσιμο εδώ
- What is ALS?, ALS-Association, διαθέσιμο εδώ
- Amyotrophic Lateral Sclerosis (ALS), John Hopkin’s Medicine, διαθέσιμο εδώ

