

Της Ζένιας Κουφοπαντελή,
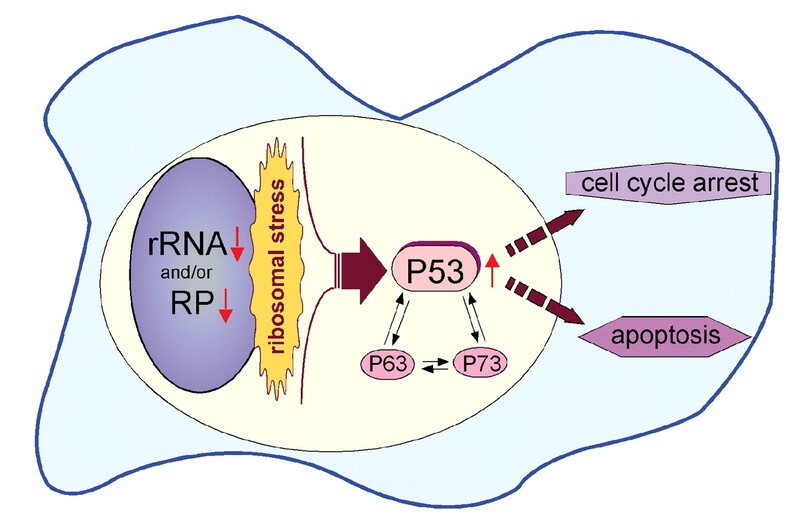
Η αναιμία Diamond-Blackfan (DBA) ήταν η πρώτη ριβοσωματοπάθεια που περιγράφηκε στον άνθρωπο και αρχικά γνωστή με τον όρο συγγενής υποπλαστική αναιμία. Πρόκειται για ένα κληρονομικό σύνδρομο ανεπάρκειας του μυελού των οστών, που υπολογίζεται ότι εμφανίζεται περίπου σε 7 στις 1.000.000 γεννήσεις. Η διάγνωση τίθεται μετά τη γέννηση κατά τους πρώτους μήνες ζωής, με διάμεση ηλικία να αποτελούν οι 3 μήνες. Η DBA έχει, επίσης, διαγνωστεί κατά τη γέννηση σε ποσοστό 13% έως 16% των περιπτώσεων. Το κύριο χαρακτηριστικό της κλασικής DBA είναι ένα ελάττωμα στην επεξεργασία του ριβοσωμικού RNA, που οδηγεί σε σταθεροποίηση της πρωτεΐνης p53 και ενεργοποίηση των στόχων του, με αποτέλεσμα τη διακοπή του κυτταρικού κύκλου και την απόπτωση.
Η νόσος χαρακτηρίζεται από ισχυρό γενετικό υπόβαθρο, καθώς οφείλεται κατά βάση σε αυτοσωμική κληρονομικότητα ή σποραδικές μεταλλάξεις. Μέχρι σήμερα έχουν εντοπιστεί μεταλλάξεις σε 20 γονίδια ριβοσωμικών πρωτεϊνών (RP), που σχετίζονται με τη DBA. Μεταξύ αυτών, έχει εντοπιστεί η μετάλλαξη στο γονίδιο RPS19, που κωδικοποιεί αντίστοιχα για την πρωτεΐνη RPS19. Η πρωτεΐνη αυτή αποτελεί βασικό συστατικό του σωματίου ματίσματος (spliceosome), διαδραματίζοντας ρόλο και στην ερυθροποιητική διαφοροποίηση και τον πολλαπλασιασμό, εκτός από τη λειτουργία της ως συστατικού του ριβοσώματος. Σε όλες τις περιπτώσεις όμως, οι μεταλλάξεις των γονιδίων RP οδηγούν σε ελαττωματική ωρίμανση του ριβοσωματικού RNA (rRNA). Επιπρόσθετα, έχει προταθεί πως η εξασθένιση της βιογένεσης των ριβοσωμάτων, λόγω μείωσης μίας RP, μπορεί να προκαλέσει ενεργοποίηση της p53. Το γεγονός αυτό έχει ως επακόλουθο την αναστολή της κυτταρικής ανάπτυξης και, κατ’ επέκταση, την απόπτωση. Σε πολλούς ασθενείς, ωστόσο, πέρα από μεταλλάξεις των γονιδίων που κωδικοποιούν RP, ορισμένοι ασθενείς εμφανίζουν ερυθροβλαστοπενία που οφείλεται σε μεταλλαγμένα γονίδια, τα οποία δεν κωδικοποιούν για RP (GATA1, EPO, ADA2). Από την άλλη, ορισμένοι ασθενείς χωρίς ιστορικό αναιμίας εκδηλώνουν παθογόνο μετάλλαξη σε γονίδιο RP, που σχετίζεται με την εμφάνιση λευχαιμίας ή με άλλες παθολογικές καταστάσεις.
Στα βρέφη, η DBA παρουσιάζεται μέσα από σημάδια αναιμίας, όπως ωχρότητα, αδυναμία ανάπτυξης και δυσκολίες στο θηλασμό κατά τη διάρκεια του θηλασμού ή και έπειτα. Εκτός από την αναιμία, διάφορες συγγενείς δυσπλασίες εκδηλώνονται στο 50% των ασθενών που πάσχουν από DBA. Σε ποσοστό ίσο με το 20% των περιπτώσεων συμβαίνει πρόωρος τοκετός, ενώ, παράλληλα, έχει καταγραφεί καθυστέρηση στην ανάπτυξη σε ορισμένες περιπτώσεις. Το κοντό ανάστημα που παρατηρείται κατά την ενδομήτρια ζωή εκδηλώνεται σε ποσοστό περίπου 22% των περιπτώσεων. Αυτό επιδεινώνεται σταδιακά, λόγω των ανεπιθύμητων επιδράσεων ποικίλων θεραπειών, όπως η θεραπεία με στεροειδή και η υπερφόρτωση σιδήρου που προκαλείται λόγω μετάγγισης. Εντοπίζονται δυσμορφίες, κυρίως στην κεφαλική περιοχή, στο 50% των ασθενών, αλλά και στα άκρα. Αυτές περιλαμβάνουν ανωμαλίες του αντίχειρα με παθογνωμονικό σύμπτωμα, τον τριγωνικό αντίχειρα. Άλλες περιοχές που συχνά προσβάλλονται, αποτελούν η ουρογεννητική οδός και η καρδιά. Επιπρόσθετες ενδείξεις για την ύπαρξη της ασθένειας σχετίζονται με:
- Ορθόχρωμη, και κατά βάση μακροκυτταρική αναιμία
- Φυσιολογικός ή ελαφρά ελαττωμένος αριθμός λευκών αιμοσφαιρίων
- Φυσιολογικός ή αυξημένος αριθμός αιμοπεταλίων
- Απουσία δικτυοερυθροκυττάρων
- Μυελός των οστών, όπου παρατηρείται εκλεκτική ανεπάρκεια των πρόδρομων ερυθροκυττάρων
Για τους πάσχοντες με DBA, κρίνεται επιτακτική μια πολύπλευρη προσέγγιση για την αντιμετώπισή της, κάτι που εξαρτάται και με την ηλικία του ασθενούς. Κατά τη γέννηση και κατά τη διάρκεια της βρεφικής ηλικίας, τα επίπεδα αιμοσφαιρίνης (Hb) μπορεί να είναι είτε φυσιολογικά είτε κάτω του φυσιολογικού, αλλά η ανάγκη για μετάγγιση εμφανίζεται πριν από την ηλικία του 1 έτους στην πλειονότητα των ασθενών. Η χορήγηση γλυκοκορτικοειδών συνίσταται για ηλικίες άνω του 1 έτους και όχι νωρίτερα, ενώ μπορεί να καθυστερήσει περαιτέρω σε παιδιά με σοβαρή ανεπάρκεια ανάπτυξης. Για τους ασθενείς που κάνουν μετάγγιση, η κύρια παρενέργεια είναι ο κίνδυνος υπερφόρτωσης με σίδηρο. Επομένως, η εφαρμογή της αποτελεσματικής και εντατικής θεραπείας χηλίωσης επιβάλλεται για τους πάσχοντες από DBA, ακόμη και σε νεαρή ηλικία. Η θεραπεία αυτή ξεκινάει συνήθως πριν την ηλικία των 2 ετών, ανάλογα με την ηλικία κατά την οποία ξεκίνησε η πρώτη μετάγγιση και με τα επίπεδα φεριτίνης. Στην ηλικία αυτή, η δεφεροξαμίνη επιλέγεται ως χηλικός παράγοντας. Τέλος, άλλες επιλογές, οι οποίες, ωστόσο, βρίσκονται υπό διερεύνηση, αποτελούν η γονιδιακή θεραπεία και η χρήση πολυδύναμων βλαστικών κυττάρων.
ΕΝΔΕΙΚΤΙΚΕΣ ΠΗΓΕΣ
- Diamond-Blackfan anemia, pubmed. Διαθέσιμο εδώ
- Συγγενή/κληρονομικά σύνδρομα μυελικής ανεπάρκειας, upatras.gr. Διαθέσιμο εδώ
- The Genetic Landscape of Diamond-Blackfan Anemia, ncbi.nlm.nih.gov. Διαθέσιμο εδώ
- RPS19 Gene – Ribosomal Protein S19, genecards.org. Διαθέσιμο εδώ
- Diamond-Blackfan anemia: a ribosomal puzzle, haematologica.org. Διαθέσιμο εδώ

