

By Frangiska Mylona,
April 17th has been established as World Hemophilia Day, since 1989, to raise awareness and understanding of hemophilia and other bleeding disorders. Hemophilia is known as the “royal” disease. Many royal families suffered from this disease, which causes blood to not clot properly. Hemophilia is a royal disease that begins with Queen Victoria sometimes referred to as the “Mother of Europe”. The Queen, in order to ensure peace throughout Europe, was intermarrying her grandchildren creating bonds with the royal families of Spain, Germany, and, even Russia. Queen Victoria was a carrier of this disease from a spontaneous mutation in her genome. One, also very known family, for having offspring with hemophilia is the Romanov family in Russia (known also as the Last Tsars) with their son Aleksei that in order to treat their child, they completely entrusted Grigori Rasputin, a mystic and self-proclaimed holy man with healing abilities that it is believed to be one of the factors leading to the overthrown and demise of the Romanov family.

As it is understood hemophilia refers to the inability of blood to clot properly or even clot at all. It is associated with diseases due to coagulation factor deficiencies. In normal conditions, hemostasis (clotting process) happens in three steps ;
● Vessel vasoconstriction; platelets release vasoconstrictive substances such as Thromboxane A2, Serotonin, and ADP.
● Formation of platelet plug; seconds after the vessel injury the VWF (Von Willebrand Factor ) is released from the endothelium of the blood vessel adhering platelets together to the exposed collagen fibers on the damaged vessel wall. By binding to the vessel wall, the platelets continue to release substances such as ADP and thromboxane A2. This leads to platelet aggregation with one another as well.
●Blood coagulation; The platelet plug is not strong enough to induce hemostasis. It needs fibrin reinforcement. Fibrin is the product of fibrinogen when the enzyme thrombin acts on it. To produce thrombin the coagulation cascade gets activated. It is called a cascade because the activation of a coagulation factor is required for the activation of the next factor in the sequence. This cascade is divided into two pathways; intrinsic and extrinsic which are based on the activation of the coagulation factors to produce thrombin. These factors are designated with Roman numerals and are twelve in number. However, they are enumerated as thirteen, but Factor VI is thought of as part of another coagulation factor.
Most coagulation factors are synthesized in the liver where Vitamin K is necessary for their production. Differences exist between intrinsic and extrinsic pathways. The intrinsic pathway is slower (can cause clotting in 1 to 6 minutes) and gets activated as soon as the blood comes in contact with collagen in the injured vessel wall. On the other hand, the extrinsic pathway is faster (can cause clotting in 15 seconds) and gets activated when there is a trauma to the blood vessel that subsequently releases tissue factor from the subendothelial cells. However, the terminal steps in both pathways are the same leading to the activation of factor X which will convert prothrombin (factor II) to thrombin (factor IIa) which will later help in fibrin formation. One significant piece of information is that calcium ions are also a coagulation factor (Factor IV) used in all steps except the first two where the body has enough calcium for these reactions.
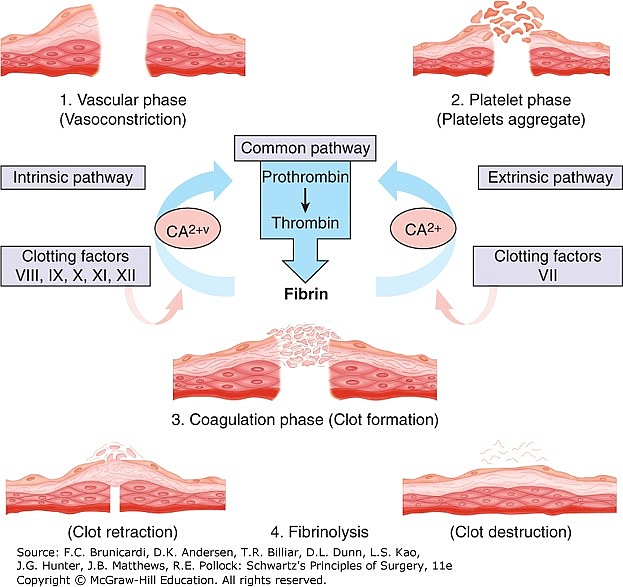
As with every process in the human body, coagulation needs to be regulated or limited. This is possible with the action of substances like Protein C, which is also produced in the liver, inactivating factors V and VIII, tissue protein factor pathway inhibitor, protein S; a plasma protein that accelerates the action of Protein C, antithrombin III which neutralizes thrombin as well as Heparin that promotes the inactivation of clotting factors and successfully suppresses fibrin. Heparin is also used as an anticoagulant to prevent thromboembolic disorders.
● Clot retraction occurs after clot formation. The clot begins to contract and this causes the fibrin strands to be pulled toward the platelets. This causes the clot serum to squeeze and, in turn, shrink the clot.
● The last step is clot dissolution. It begins shortly after the clot is formed where plasminogen gets activated. With the aid of a very powerful activator known as tPA (tissue plasminogen activators) that will convert plasminogen to plasmin to digest fibrin causing the clot to dissolve. Interestingly, several t-PAs are produced by biotechnological methods such as recombinant DNA technology to treat acute myocardial infarction and pulmonary embolism.
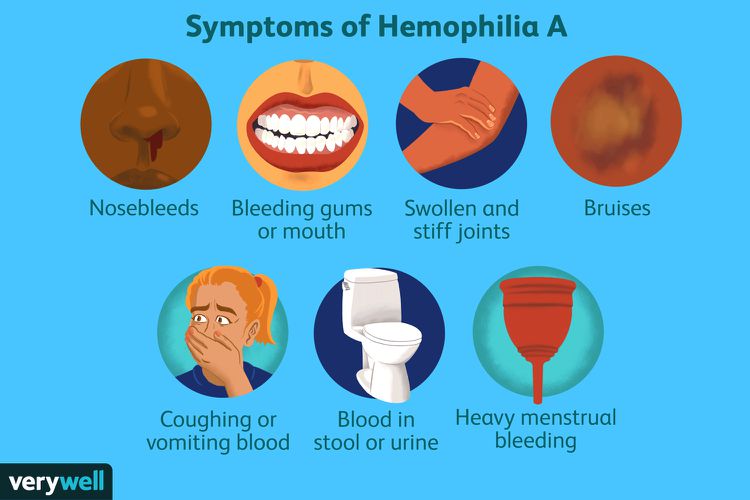
Hemophilia can be caused by congenital and acquired disorders. When it comes to congenital diseases, the two most common forms are Hemophilia type A and type B, both of which are clinically similar. Hemophilia A refers to factor VIII deficiency affecting 1 in 5000 male live births. It is inherited in an X-linked recessive manner, which means that it will primarily affect males. However, there is 30% of newly diagnosed cases without family history suggest that new mutations can arise in the factor VIII gene.
There are several forms of this type of hemophilia from mild to severe depending on whether the patient produces factor VIII in insufficient quantities or a defective form.
In mild to moderate hemophilia type A, excessive bleeding occurs after local trauma or after surgery like dental procedures. It is possible not to detect the mild form in childhood. However, in the severe form, spontaneous and severe bleeding occurs in childhood affecting the soft tissues, gastrointestinal tract, hip, elbow, and ankle joints especially when a child begins to walk. Without proper treatment, chronic bleeding in the joints can turn into fibrosis resulting in major disabilities.
Treatment usually requires the prevention of trauma as well as the avoidance of specific drugs like aspirin and NSAIDs (nonsteroidal anti-inflammatory drugs ) that interfere with the platelets. Among other therapeutic methods and prophylactic measures, Factor VIII replacement therapy is also available. The hope is that hemophilia A may be cured by gene replacement therapy based on new biotechnology-based therapeutic methods, such as cloning of the factor VIII gene. A prenatal diagnosis and carrier detection are recommended if there is a family history of hemophilia.
Similarly, hemophilia B is caused by factor IX deficiency (also known as the Christmas factor) by mutations in the factor IX gene. It occurs in approximately 1 to 20.000 people accounting for 15% of hemophilia patients. It is also inherited in an X-linked recessive manner affecting mainly males. It is the type of hemophilia that affected Queen Victoria’s descendants. As with hemophilia type A, it manifests in mild, moderate, and severe types. Treatment and prophylactic methods remain the same as hemophilia type A, including plasma-derived factor concentrates and recombinant factor concentrates, as well as preventing trauma.

Particular attention is given to Von Willebrand disease. It is the most common inherited coagulopathy, affecting almost 1% to 2% of the population. It is associated closely with hemophilia type A since it is caused by defects involving the factor VIII-VWF complex. One of its main functions is to act as a carrier for factor VIII and stabilize factor VIII in circulation, preventing proteolysis. As many as 20 variants of this disease have been identified and divided into three types. Types 1 and 3 are linked with reduced VWF levels while type 2 is characterized by VWF defects. In terms of inheritance types 1 and 2 are inherited in an autosomal dominant way while type 3 is an autosomal recessive disorder. Symptoms include spontaneous and prolonged bleeding from the nose, mouth, and gastrointestinal tract as well as heavy menstrual flow in women. Types 1 and 2 are mild and do not require treatment, whereas type 3 is severe and causes life-threatening gastrointestinal bleeding. However, most cases are mild and no treatment is required apart from avoiding aspirin.
As mentioned earlier, coagulation disorders can also be acquired. Proper coagulation and production of coagulation factors stem from the liver since it is the site of production of many coagulation factors such as V, VII, IX, X, XI, XII, prothrombin, and fibrinogen. Factors II, VII, IX, X, and prothrombin require Vitamin K to function properly.
Due to liver diseases, clotting factors are less synthesized, resulting in easier bleeding. When there is a Vitamin K deficiency the liver produces inactive clotting factors. Vitamin K deficiency is linked with intestinal problems since it is produced by intestinal bacteria or by disorders regarding its absorption usually caused by liver or gallbladder diseases since, Vitamin K, is a fat-soluble protein that in order to be absorbed, it requires bile salts. Commonly, Vitamin K deficiency occurs in newborns because they do not possess yet normal intestinal flora as well as in patients treated with antibiotics destroying the intestinal flora. Other acquired clotting disorders can be secondary due to anticoagulant drugs like Herapin or conmarine anticoagulants like Warfarin.
Moreover, hemophilia type A can be an acquired disorder. It is a rare disorder resulting in spontaneous bleeding in individuals with no history of bleeding disorders. It is believed to be caused by spontaneous inhibition of clotting factor VIII by autoantibodies and is usually associated with other autoimmune conditions. The primary approach to treat acquired hemophilia types is to manage the underlying diseases.
Apart from drug therapies, improving nutritional habits can be of great assistance to people suffering from coagulation disorders. For example incorporating foods with iron because people with hemophilia lose iron along with blood, increasing the whole grain intake, switching to a low- fat diet by choosing alternative cooking methods than frying, and paying attention when it comes to supplements like Vitamin E, although it is involved in red blood cell production, it can increase the risk for bleeding disorders. However, other supplements that seem helpful are Vitamin K, Vitamin C, Vitamin B12, and folic acid. Further engagement types include the reduction of portion size and paying for ‘’hidden‘’ calories in foods, soft drinks, and juices.
Consequently, diseases do not discriminate based on whether a person has royal blood or not. Everyone can be affected by hemophilia, regardless of their status or characteristics. The disease is characterized by prolonged bleeding and clotting inability. In the past, the disease was even more restrictive than it is now, with the fear of falling or getting injured. A process often taken for granted by many people regarding the treatment of wounds and the clotting process, it can pose a major problem for people with hemophilia. World Hemophilia Day is dedicated to raising awareness of possible risks, helping people in need, finding effective and newer ways of treatment as well as honoring the memory of those who have passed away from this disease.
References
- Porth’s Essential of Pathophysiology; 5th edition by Tommie L.Norris and contributing author; Rupa Lalchandani Tuan, PhD
- Nutrition in hemophilia. hemophilianewstoday.com. Available here

