

Της Κατερίνας Μπουμπούλη,
Ο ανθρώπινος οργανισμός συχνά παρομοιάζεται με ένα πολύπλοκο σύστημα, μια ορθότατη σύγκριση αν αναλογιστεί κανείς ότι αποτελείται από πλήθος οργάνων, που το καθένα συγκροτείται από τεράστιο αριθμό κυττάρων. Το σύστημα αυτό καλείται διαρκώς και ακατάπαυστα να βρίσκεται υπό απόλυτο έλεγχο και να ολοκληρώνει κάθε διεργασία αλάνθαστα και ταχύτατα. Προκειμένου να αποφευχθούν πιθανές καθυστερήσεις και να διαταραχθεί η ισορροπία, το σώμα μας παράγει ένζυμα. Ως ένζυμα ορίζονται οι πρωτεΐνες που συμβάλλουν στην επιτάχυνση του μεταβολισμού και των χημικών αντιδράσεων του οργανισμού μας. Ουσιαστικά, διασπούν ουσίες, σχηματίζοντας νέες. Προφανώς, υπάρχει μεγάλη ποικιλία αυτών, εξειδικευμένων για αντιδράσεις και υποστρώματα.
Η νόσος του Fabry, αποτελεί μια περίπτωση έλλειψης ή παντελούς απουσίας ενός ενζύμου, της α-γαλακτοσιδάσης Α (GLA), που διαταράσσει τον γλυκοσφιγγολιπιδικό μεταβολισμό και προκαλείται έπειτα από μεταλλαγές στο γονίδιο που κωδικοποιεί την α-γαλακτοσιδάση Α. Φυσιολογικά, τα λυσοσώματα λειτουργούν ως η κύρια μεταβολική οδός των κυττάρων, αφού με τη βοήθεια των ενζύμων, διασπούν σύμπλοκα σακχάρων και λιπιδίων, δομές γνωστές ως γλυκολιπίδια. Παρομοίως, η GLA είναι υπεύθυνη για τη διάσπαση του σφαιροτριαζυλοκεραμιδίου (GL3/Gb3), της απακυλιωμένης του μορφής, καθώς και άλλων συναφών γλυκολιπιδίων, αφαιρώντας το τελικό σάκχαρο γαλακτόζης αυτών των μορίων. Η έλλειψη ενζύμου οδηγεί στην ατέρμονη συσσώρευση GL3/Gb3 στα κύτταρα του σώματος, με αποτέλεσμα την εμφάνιση κυτταρικών και οργανικών δυσλειτουργιών, που με τη σειρά τους επηρεάζουν μικρά αιμοφόρα αγγεία, την καρδιά και τα νεφρά.
Συνεπώς, η νόσος ανήκει στις διαταραχές λυσοσωμικής αποθήκευσης. Θεωρείται σπάνια, ενώ κληρονομείται ακολουθώντας φυλοσύνδετο μοτίβο. Πιο συγκεκριμένα, το γονίδιο που κωδικοποιεί το ένζυμο ανιχνεύεται στο Χ χρωμόσωμα, δηλαδή σε ένα από τα δύο φυλετικά χρωμοσώματα. Έτσι, ένα αρσενικό άτομο αρκεί να έχει τη μετάλλαξη μόνο στο ένα χρωμόσωμα, αφού διαθέτει ένα Χ και ένα Υ. Αντίθετα, ένα θηλυκό άτομο με δύο Χ χρωμοσώματα, εμφανίζει πιο ήπια συμπτώματα της ασθένειας σε περίπτωση μετάλλαξης του ενός χρωμοσώματος και σοβαρότερα, εάν η συγκεκριμένη μετάλλαξη βρίσκεται και στα δύο.
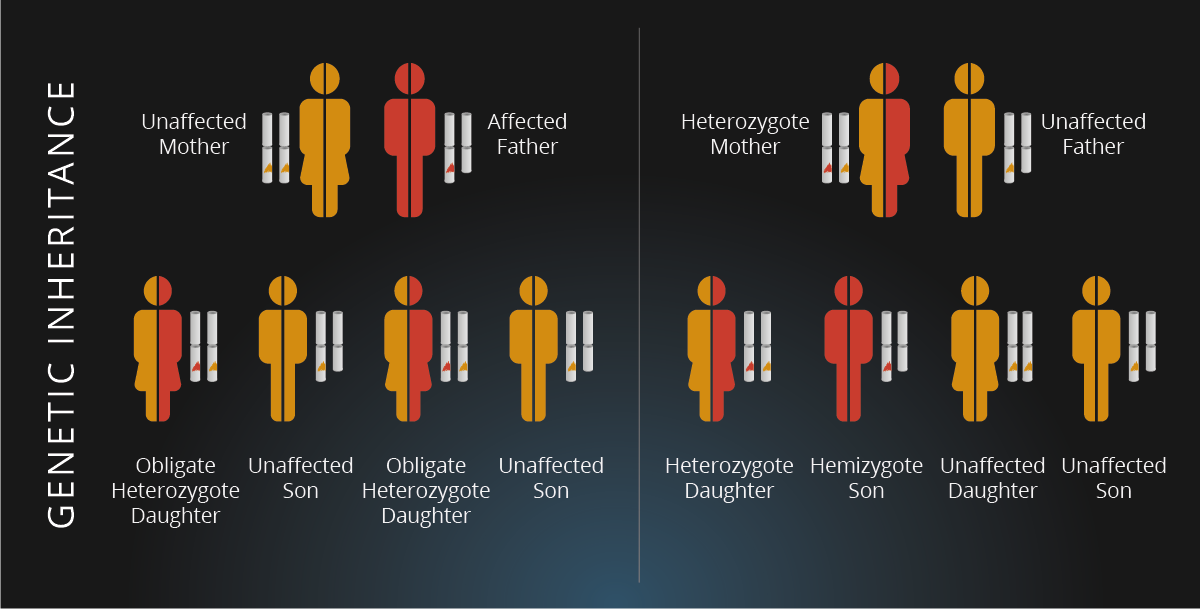
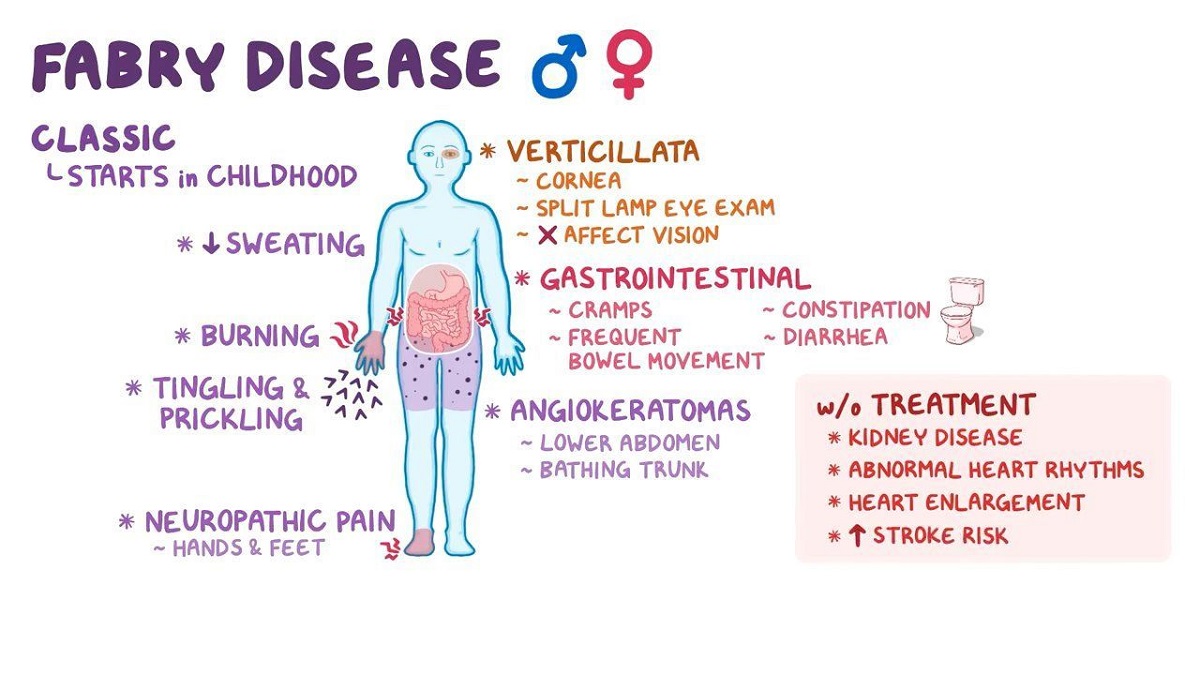
Γενικά, υπάρχουν δύο κύριοι φαινότυποι της ασθένειας, οι οποίοι οδηγούν σε νεφρική ανεπάρκεια ή/και καρδιακά προβλήματα. Η νόσος Fabry τύπου Ι ή κλασική, αφορά άντρες με ελάχιστα ή καθόλου λειτουργικό γονίδιο, του οποίου η λειτουργία υπολογίζεται πως είναι μικρότερη από το 3% της φυσιολογικής. Εξαιτίας αυτού, παρατηρείται έντονη συσσώρευση GL3/Gb3, καθώς και των υπόλοιπων σχετικών γλυκολιπιδίων σε τριχοειδή και αιμοφόρα αγγεία, προκαλώντας τα κύρια συμπτώματα της νόσου ήδη από την παιδική ή την εφηβική ηλικία. Αρχικό σύμπτωμα της ασθένειας είναι η ακροπαραισθησία, που εκδηλώνεται με πόνο που γίνεται αισθητός σαν κάψιμο στα χέρια και στα πόδια και πυροδοτείται από την άσκηση, το άγχος, τον πυρετό και την κούραση. Στα συμπτώματα περιλαμβάνεται και το αγγειοκεράτωμα, το οποίο εμφανίζεται σαν κοκκινωπά έως σκούρα μπλε εξανθήματα του δέρματος, ειδικά μεταξύ των γοφών και των γονάτων, αλλά και στην ομφαλική περιοχή και στα αντρικά γεννητικά όργανα. Συμπληρωματικά, συχνά παρουσιάζεται ανιδρωσία ή υποϊδρωσία, δηλαδή παντελής απουσία ή μείωση εφίδρωσης, γαστρεντερικές δυσλειτουργίες συνοδευόμενες από κοιλιακό άλγος, κράμπες και συχνές κενώσεις, καθώς και δυστροφία κερατοειδούς χωρίς να επηρεάζει την όραση.
Σε μεγαλύτερης ηλικίας άτομα που πάσχουν από τον κλασικό τύπο της νόσου, εμφανίζονται και σοβαρότερα προβλήματα. Λόγω συστηματικής εναπόθεσης γλυκολιπιδίων, αφού αυτά δε μεταβολίζονται, κυρίως στην καρδιά, προκαλείται αρρυθμία, υπερτροφία της αριστερής κοιλίας, αλλά και υπερτροφική μυοκαρδιοπάθεια. Αντίστοιχες δυσλειτουργίες παρατηρούνται και στα νεφρά, όπως προοδευτική πρωτεϊνουρία και νεφρική ανεπάρκεια. Τέλος, οι εγκεφαλοαγγειακές νόσοι με παροδικές ισχαιμικές κρίσεις και τα εγκεφαλικά επεισόδια θεωρούνται επακόλουθα της ασθένειας. Γενικά, η νόσος του Fabry τύπου Ι κληρονομείται σε 1 στα 40.000 άτομα, ενώ η μέση ηλικία θανάτου υπολογίζεται στα 40 έτη. Ωστόσο, το προσδόκιμο ζωής αυξάνεται σημαντικά έπειτα από θεραπείες υποκατάστασης νεφρών, όπως αιμοκάθαρση και μεταμόσχευση, καθώς και ενζυμική θεραπεία.
Αντίθετα, ο τύπος ΙΙ της ασθένειας, ή αλλιώς ο τύπος μεταγενέστερης έναρξης, χαρακτηρίζεται από μειωμένη δραστηριότητα ενζύμου που, όμως, δεν προκαλεί συσσώρευση του GL-3/Gb3 σε τριχοειδή και μικρά αιμοφόρα αγγεία. Επίσης, δεν εμφανίζονται τα αρχικά συμπτώματα (ακροπαραισθισία, υποϊδρωσία, ανιδρωσία, αγγειοκεράτωμα, δυστροφία κερατοειδούς) σε μικρή ηλικία, αλλά ο ασθενής απολαμβάνει μια κανονική παιδική και εφηβική ηλικία. Ωστόσο, μεταξύ της τρίτης και της έβδομης δεκαετίας της ζωής, του παρουσιάζει κάποια νεφρική ή καρδιακή νόσο. Η ύπαρξη της ασθένειας μπορεί να διαπιστωθεί μόνο με ενζυμικό έλεγχο. Ο συγκεκριμένος τύπος της νόσου φαίνεται να εμφανίζεται 5 έως 10 φορές συχνότερα από τον κανονικό τύπο Ι.
Όπως αναφέρθηκε, το μοτίβο κληρονόμησης στις γυναίκες διαφέρει αρκετά από αυτό των αντρών. Οι ετερόζυγες γυναίκες που πάσχουν από τον κλασικό τύπο της νόσου Fabry, δηλαδή που έχουν μόνο ένα μεταλλαγμένο γονίδιο, δεν εμφανίζουν πάντα σοβαρά συμπτώματα, όπως συνηθίζεται στους άντρες, αλλά μπορεί να είναι και ασυμπτωματικές. Παρομοίως, και στη νόσο τύπου ΙΙ, μπορεί να μην υποφέρουν από τα συμπτώματα που προαναφέρθηκαν ή μπορεί να εμφανίσουν νεφρικά και καρδιακά προβλήματα σε μετέπειτα στάδια της ζωής τους. Παρόλα αυτά, περίπου το 90% των περιπτώσεων τύπου Ι υποφέρει από δυστροφία κερατοειδούς.
Τα ένζυμα, λοιπόν, διαδραματίζουν καθοριστικό ρόλο στη σωστή λειτουργία και στη διατήρηση της ισορροπίας του οργανισμού. Η μετάλλαξη ενός μόνο γονιδίου μπορεί να επιφέρει συνέπειες που θα οδηγήσουν το άτομο ακόμη και σε θάνατο. Ωστόσο, η πρόοδος της βιολογίας, της ιατρικής και της χημείας, αλλά και η συνεργασία των επιστημών προλαμβάνει και συμβάλλει στην εξασφάλιση καλύτερων συνθηκών ζωής.
ΕΝΔΕΙΚΤΙΚΕΣ ΠΗΓΕΣ
- Fabry Disease, National Organization of Rare Disorders. Διαθέσιμο εδώ
- Fabry Disease, Medline Plus. Διαθέσιμο εδώ
- Enzymes, Cleveland Clinic. Διαθέσιμο εδώ

